
Making a MASH Hit: PNPLA3 and the Rise of Genotype-Driven Therapies
•February 16, 2026
0

Why It Matters
Targeting PNPLA3 I148M attacks the root cause of lipid accumulation, offering a potential disease‑modifying solution for MASH and validating genotype‑driven strategies in metabolic disorders.
Key Takeaways
- •PNPLA3 I148M drives lipid droplet accumulation in MASH
- •RNAi and ASO therapies now in clinical development
- •Pfizer's covalent degrader targets mutant protein for proteolysis
- •Novartis PPI inhibitor disrupts PNPLA3‑CGI58 interaction
- •Success could validate genotype‑driven drug discovery for metabolic disease
Pulse Analysis
The surge of GLP‑1R agonists has highlighted the unmet need for disease‑modifying treatments in metabolic dysfunction‑associated steatohepatitis (MASH). While many metabolic pathways—FGF21, DGAT2, HSD17B13, PPAR agonists—have stumbled in late‑stage trials, the PNPLA3 I148M variant stands out for its strong human genetic association with severe liver disease. Carriers of the homozygous mutation exhibit markedly higher risk of fibrosis and inflammation, making PNPLA3 a rare example where a single amino‑acid change directly drives pathology, and thus a prime candidate for precision therapeutics.
RNA‑targeting platforms have capitalized on this insight by silencing the mutant transcript rather than inhibiting enzymatic activity. Arrowhead’s GalNAc‑conjugated siRNA ARO‑PNPLA3 and AstraZeneca/Ionis’ GalNAc‑ASO AZD2693 both aim to reduce hepatic PNPLA3 expression, leveraging liver‑specific delivery to achieve potent knock‑down with minimal systemic exposure. Early clinical data suggest favorable safety profiles, underscoring the advantage of transcript reduction for a target where protein accumulation, not catalytic excess, fuels disease. These programs illustrate how antisense and RNAi technologies can translate genetic validation into actionable therapeutic strategies.
Small‑molecule innovation is catching up, as evidenced by Pfizer’s covalent degrader PF‑07853578 and Novartis’ reversible PPI inhibitor. The former employs a warhead to covalently bind the mutant’s catalytic serine, flagging it for proteasomal degradation, while the latter disrupts the PNPLA3‑CGI58 interaction that impairs lipid hydrolysis. Both approaches confront the challenge of selectively targeting the mutant without affecting the wild‑type enzyme, a hurdle that has limited traditional inhibition attempts. Success across these modalities would not only deliver a first‑in‑class MASH therapy but also signal a new era where genotype‑defined targets guide drug discovery for complex metabolic diseases.
Making a MASH Hit: PNPLA3 and the Rise of Genotype-Driven Therapies
The rise of GLP‑1R agonists and the recent approval of semaglutide for MASH have reignited interest in drug targets for severe liver diseases. MASH remains one of the most unforgiving indications in the clinic; multiple mechanisms with promising pre‑clinical biology—including FGF21, DGAT2, HSD17B13, PPAR agonism, among others—have led to disappointing clinical outcomes. Against this background, an important question must be asked: beyond metabolic modulation, which targets can address the underlying drivers of disease, and what has held progress back? One target attracting increased attention is the lipid serine hydrolase PNPLA3, specifically the genetically validated I148M mutant.
PNPLA3 Steps into the Spotlight
Surveying the Drug Hunter Curated Patent Search, PNPLA3 (also known as adiponutrin) emerged as a genetically validated target in metabolic liver disease, particularly for MASLD/MASH. Unlike many therapeutic targets, the pathology of the I148M mutant is driven by its accumulation on hepatic lipid droplets, which disrupts normal triglyceride metabolism by other enzymes leading to fat retention and increasing the risk for inflammation, fibrosis, and MASH disease progression. Homozygous PNPLA3(I148M) carriers show a markedly elevated risk of MASH, providing strong human genetic validation for this target and enabling patient stratification. Importantly, genetic evidence suggests that reducing PNPLA3 levels is well tolerated, making it an attractive drug target.
From Large Molecules to Small Molecules
The strongest clinical data in support of this mechanism so far has come from RNA‑targeting approaches. Both siRNA and ASO (antisense oligonucleotide) approaches have entered clinical development with the goal of reducing hepatic PNPLA3(I148M) expression.
Arrowhead’s GalNAc‑conjugated siRNA ARO‑PNPLA3 (JNJ‑75220795) was designed to lower mutant protein levels by reducing hepatic PNPLA3(I148M) transcripts, while AstraZeneca and Ionis advanced the GalNAc‑ASO AZD2693 with a similar strategy. These approaches directly address the central biological hypothesis: the pathophysiology of the PNPLA3(I148M) protein is driven by expression levels rather than excessive catalytic activity of the protein.
This distinction helps explain why small molecules have struggled against this target; inhibition of the PNPLA3 enzymatic function should not alter disease pathology if the mutant protein continues to accumulate. As a result, RNA‑targeting approaches have been the most straightforward way to translate genetic insight into therapeutic intervention.
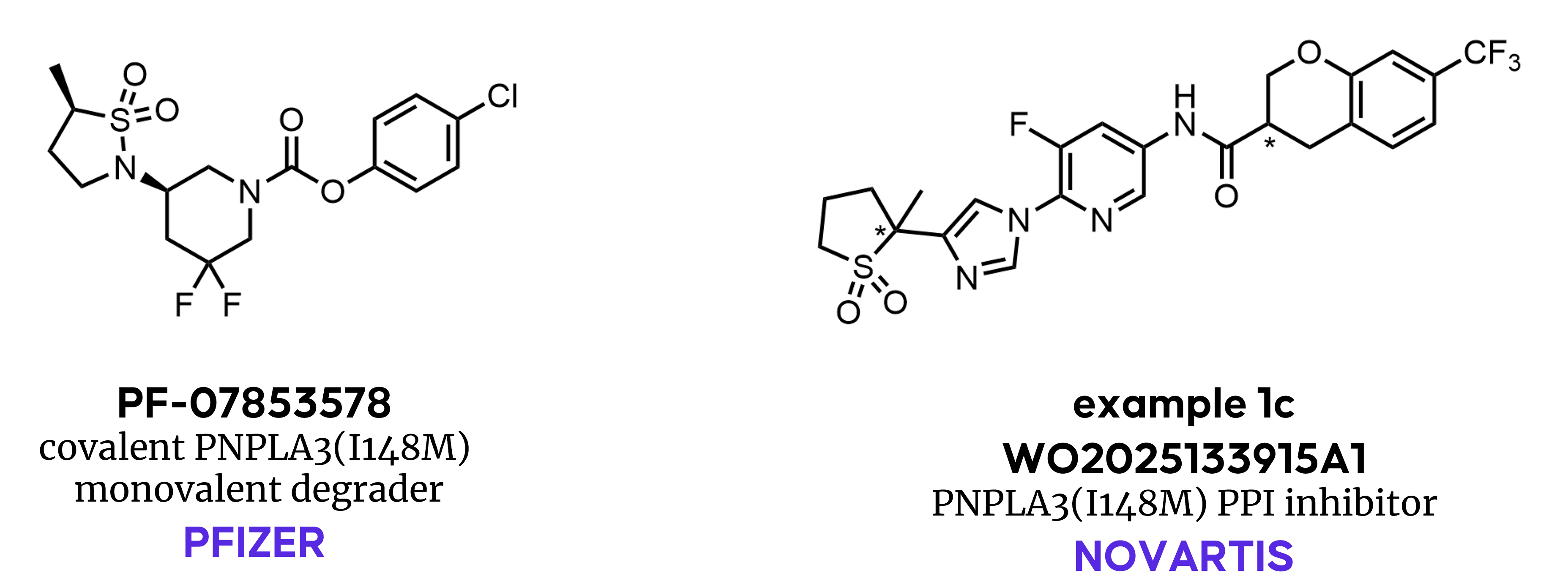
A Surprising Modality: PNPLA3 Monovalent Degrader
Recent efforts targeting PNPLA3(I148M) demonstrate advances in the scope of small‑molecule modalities. Pfizer's small‑molecule program targeting PNPLA3(I148M) represents an attempt to tackle historically difficult targets using modern drug discovery.
Our patent search highlighted compound 11 from Pfizer’s patent application WO2024084363A1, which was later disclosed as PF‑07853578 at the 2025 Spring ACS in San Diego. This compound covalently engages the catalytic serine of PNPLA3(I148M), leading to a non‑traditional, monovalent degradation mechanism that promotes removal of the mutant protein from lipid droplets. The discovery campaign illustrates the challenges inherent to this mechanism:
-
The SAR observed by fine‑tuning the warhead reactivity to achieve selectivity over other serine hydrolases did not correlate with traditional kinact/KI metrics.
-
No structural information was available to guide the optimization.
-
Wild‑type PNPLA3 is turned over relatively rapidly, whereas the I148M mutant appears to escape ubiquitylation and proteasomal degradation, leading to accumulation in hepatocytes.
Achieving sufficient target reduction while avoiding off‑target inhibition of other serine hydrolases required a delicate balance of potency, selectivity, and exposure.
The discovery effort stands as an example of how far small‑molecule modalities have evolved, moving beyond simple inhibition toward targeted modulation of proteostasis.
What’s Next?
Highlighting continued industry interest in small‑molecule modalities for this target, Novartis has recently disclosed a patent application (WO2025133915A1) covering a distinct class of molecules. These compounds appear to reversibly bind to mutant PNPLA3(I148M), alleviating the effects of its accumulation. The patent claims that the binding interferes with a protein‑protein interaction between PNPLA3(I148M) and a key activator of lipid hydrolysis, CGI58, preventing its sequestration and restoring lipid metabolism by other hydrolases.
With RNA knockdown, ASOs, covalent degraders, and PPI inhibitors now emerging, PNPLA3(I148M) has become a proving ground for genotypic drug discovery in MASH. Clinical outcomes across these modalities will be closely watched, not only for what they reveal about PNPLA3, but also for what they signal about the future of genetically defined drivers in complex metabolic diseases.
0
Comments
Want to join the conversation?
Loading comments...