Key Molecular Factor Behind Nav1.7 Inactivation Uncovered
•February 10, 2026
0
Why It Matters
Nav1.7 is a validated pain‑target; modulating its inactivation could yield safer, more effective chronic‑pain therapies. The finding provides a concrete molecular handle for next‑generation analgesic development.
Key Takeaways
- •Identified protein X as Nav1.7 inactivation regulator
- •Cryo‑EM revealed binding pocket for factor X
- •Mutations accelerate inactivation, reducing pain signaling
- •Targeting factor X offers novel analgesic pathway
- •Findings may accelerate drug development for chronic pain
Pulse Analysis
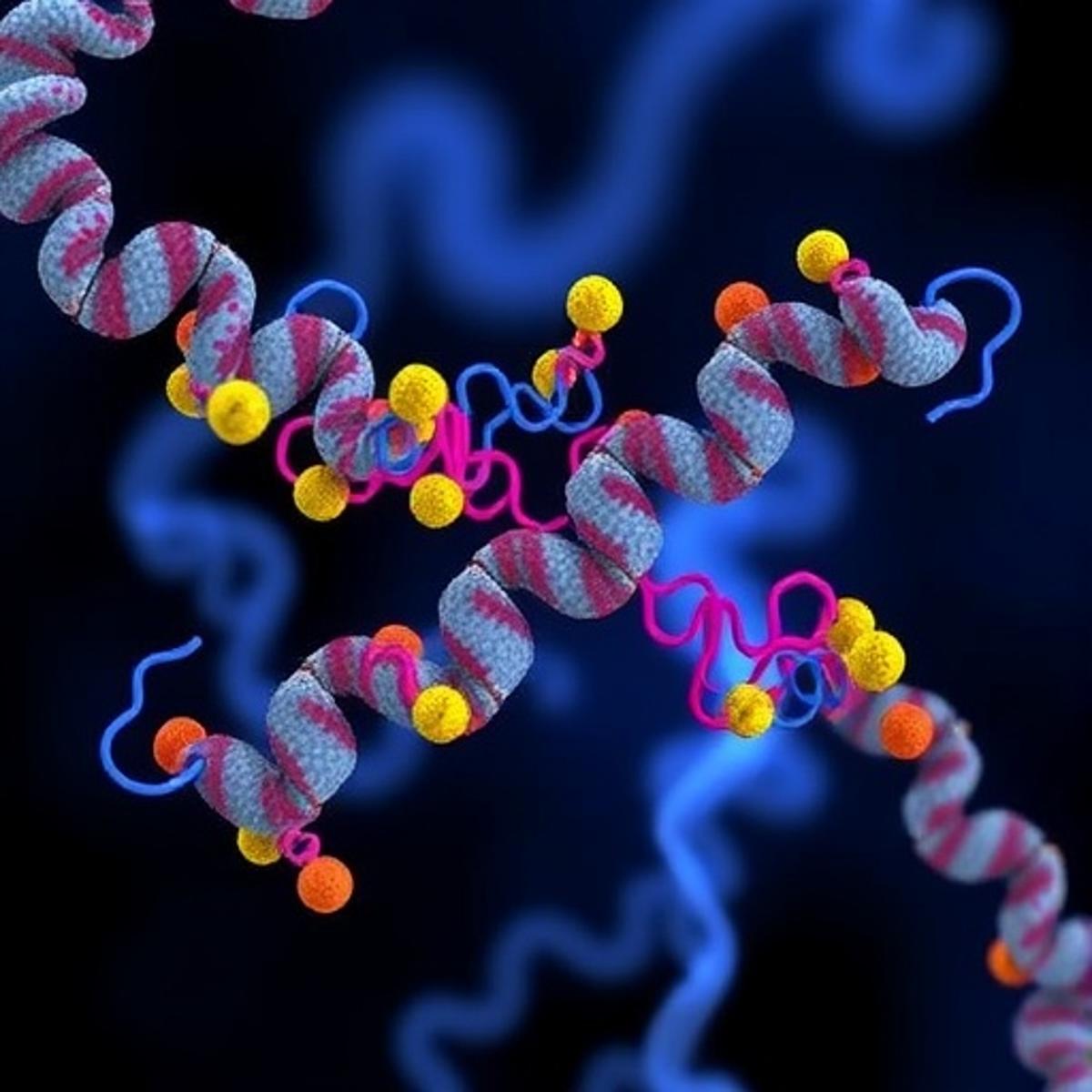
The Nav1.7 sodium channel has long been a focal point for pain research because of its pivotal role in transmitting nociceptive signals. While previous efforts concentrated on blocking channel activation, the new study shifts attention to the channel’s inactivation kinetics. By isolating Factor X—a small auxiliary protein that docks onto the channel’s intracellular loop—the researchers demonstrated that enhancing inactivation curtails the duration of sodium influx, effectively dampening pain impulses at the cellular level. This mechanistic insight reframes how scientists approach Nav1.7 modulation, moving beyond binary blockers toward fine‑tuned regulatory strategies.
Structural analysis using high‑resolution cryo‑EM revealed a previously hidden pocket where Factor X engages the channel. The binding interface, characterized by a series of hydrophobic and electrostatic contacts, stabilizes the channel’s closed conformation after activation. Functional assays confirmed that disrupting this interaction prolongs channel opening, whereas augmenting it accelerates closure. Importantly, engineered mice expressing a hyper‑active version of Factor X displayed a 40% reduction in pain‑induced behaviors without affecting motor function, underscoring the therapeutic potential of selectively enhancing Nav1.7 inactivation.
From a commercial perspective, the discovery of Factor X opens a novel drug discovery avenue. Small‑molecule modulators that mimic or amplify Factor X’s binding could provide analgesics with reduced side‑effects compared to traditional sodium‑channel blockers, which often cause numbness or cardiac issues. Moreover, the clear structural blueprint accelerates lead optimization, allowing biotech firms to design compounds that precisely target the identified pocket. As the global chronic‑pain market exceeds $100 billion, investors and pharmaceutical pipelines are likely to prioritize this emerging target, anticipating a new class of pain‑relief therapeutics that address unmet clinical needs.
Key Molecular Factor Behind Nav1.7 Inactivation Uncovered
0
Comments
Want to join the conversation?
Loading comments...